Immunodeficiency is the failure of the immune system to protect against disease or malignancy. Primary Immunodeficiency is caused by genetic or developmental defects in the immune system. These defects are present at birth but may show up later on in life. Secondary or acquired immunodeficiency is the loss of immune function as a result of exposure to disease agents, environmental factors, immunosuppression, or aging.
SECONDARY (ACQUIRED) IMMUNODEFICIENCIES
Immunodeficiencies associated with infections
Bacterial, viral, protozoan, helminthic and fungal infections may lead to B cell, T cell, PMN and macrophage deficiencies. Most prominent among these is acquired immunodeficiency syndrome (AIDS). Secondary immunodeficiencies are also seen in malignancies.
Immunologic abnormalities in the AIDS
All acquired immunodeficiencies have been outdone by AIDS that is caused by Human Immunodeficiency Virus (HIV)-1. This virus was first discovered in 1981 and the patients exhibited fungal infections with opportunistic organisms such as Pneumocystis carinii and in other cases, with a skin tumor known as Kaposi’s sarcoma. There are two major types of HIV: HIV-1 and 2, the former being the strain frequently found in North America. HIV is spread through sexual intercourse, infected blood and body fluids as well as from mother to
offspring. HIV, which was discovered in 1983, is a retrovirus with RNA that is reverse transcribed to DNA by reverse transciptase (RT) following entry into the cell. The DNA is integrated into the cell genome as a provirus that is replicated along with the cell. HIV-1 does not replicate in most other animals but infects chimpanzees although it does not induce AIDS in them. Severe combined immunodeficient mice (SCID) reconstituted with human lymphocytes can be infected with HIV-1. The HIV-1 virion consists of a viral envelope made
up of the outer lipid bilayer of the host cell in which are embedded glycoproteins composed of the transmembrane gp41 along with the associated gp120. The gp120 binds the CD4 expressed on host cells. Within the viral envelope is the viral core or nucleocapsid consisting of a layer of matrix protein composed of p17 and an inner capsid made up of p24. The viral genome consists of two single stranded RNA molecules associated with two RT molecules as
well as other enzymes including a protease and an integrase.
Replication cycle and targets of therapy
The virus attaches to the CD4 molecule on Th cells, monocytes and dendritic cells through the gp120 of HIV. For HIV infection, a co-receptor is required. The co-receptor is a chemokine receptor such as CXCR4 or CCR5. CCR5, expressed predominantly on macrophages, and CXCR4 on CD4+ T cells serve as coreceptors for HIV infection. After the fusion of HIV envelope and the host membrane, the nucleocapsid enters the cell. The RT synthesizes viral DNA which is transported to the nucleus where it integrates with the cell
DNA in the form of a provirus. The provirus can remain latent till the cell is activated when the provirus also undergoes transcription. Virions, consisting of the transcribed viral RNA and proteins, are produced. These bud out of the host cell membrane from where they acquire the envelope. Thus, therapeutic agents have been developed that target viral entry and fusion, as well as serve as RT, protease and integrates inhibitors. Highly active anti-retroviral therapy
is a cocktail of 3 or more such agents.
Immunological Changes
The virus replicates rapidly and within about two weeks the patient may develop fever. The viral load in the blood increases significantly and peaks in two months, after which there is a sudden decline because of the latent virus found in germinal centers of the lymph nodes. CTL develop very early whereas antibodies can be detected between 3 – 8 weeks. The CTL killing of
of Th cells around 4 – 8 weeks leads to a decrease in CD4+ T cells. When the CD4+ T cell count decreases below 200 per cubic mm, full blown AIDS develops.
Immunotherapy
There are several barriers to development of an effective HIV vaccine.
Attenuated vaccine may induce the disease
CD4+ T cells may be destroyed by the vaccine
Antigenic variation of HIV
Low immunogenicity of the virus by downregulation of MHC molecules
Lack of animal models
Lack of in vitro tests
The following reagents have been considered in developing vaccines:
Immunization with deletion mutants to reduce pathogenicity
Vaccination with recombinant proteins
Gene encoding proteins introduced into virus vectors may be used for
vaccination
Chemokines that compete for the co-receptors
IL-2 to boost the Th cells.
Immunodeficiencies associated with aging
These include a progressive decrease in thymic cortex, hypo-cellularity of and reduction in the size of thymus, a decrease in suppressor cell function and hence an increase in auto-reactivity, a decrease in CD4 cells functions. By contrast B cells functions may be somewhat elevated.
Immunodeficiencies associated with malignancies and other diseases
B cell deficiencies have been noted in multiple myeloma, Waldenstrom’s macroglobulinemia, chronic lymphocytic leukemia and well differentiated lymphomas. Hodgkin’s disease and advanced solid tumors are associated with impaired T-cell functions. Most chemotherapeutic agents used for treatment of malignancies are also immunosuppressive.
Other conditions in which secondary immunodeficiencies occur are sickle cell anemia, diabetes mellitus, protein calorie malnutrition, burns, alcoholic cirrhosis, rheumatoid arthritis, renal malfunction, etc.
PRIMARY IMMUNODEFICIENCIES
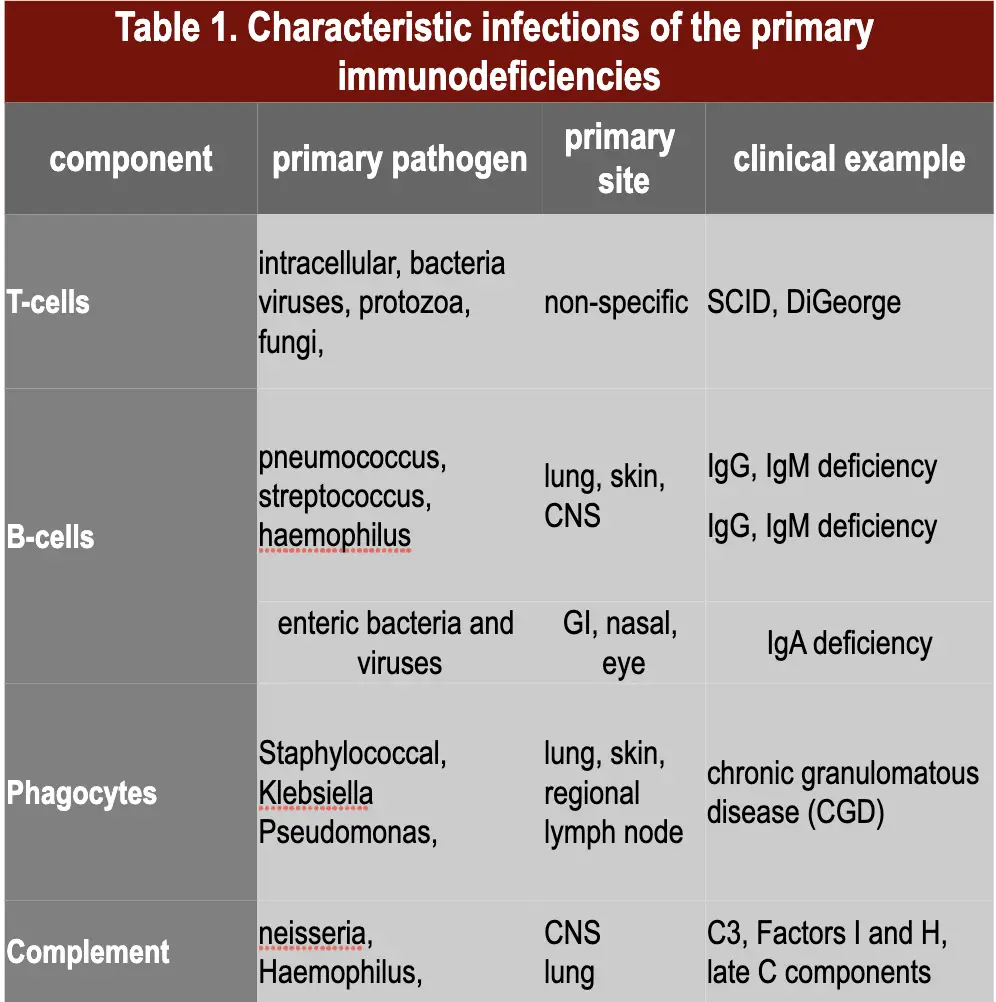
Primary immunodeficiencies are inherited defects of the immune system (figure 1). These defects may be in the specific or non-specific immune mechanisms. They are classified on the basis of the site of lesion in the developmental or differentiation pathway of the immune system. Individuals with immunodeficiencies are susceptible to a variety of infections and the
type of infection depends on the nature of immunodeficiency (Table 1).

SPECIFIC IMMUNE SYSTEM
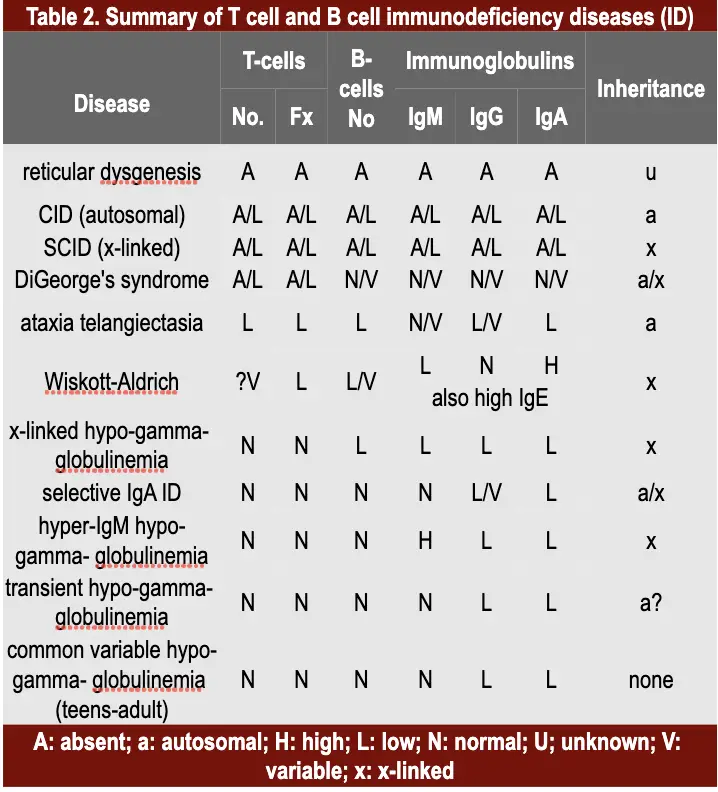
There are a variety of immunodeficiencies which result from defects in stem cell differentiation and may involve T-cells, B-cells, and/or immunoglobulins of different classes and subclasses (Table 2).
A defect in the early hematopoiesis which involves stem cells results in reticular dysgenesis that leads to general immune defects and subsequent susceptibility to infections. This condition is often fatal but very rare.
Lymphoid lineage immunodeficiency
If the lymphoid progenitor cells are defective, then both the T and B cell lineages are affected and result in the severe combined immunodeficiency (SCID). Infants suffer from recurrent infections especially by opportunistic micro-organisms (bacterial, viral, mycotic and protozoan infections).
In about 50% of SCID patients, the immunodeficiency is x-linked whereas in the other half the deficiency is autosomal. Both are characterized by an absence of T cell and B cell immunity and absence (or very low numbers) of circulating T and B lymphocytes. Thymic shadows are absent on X-rays.
The x-linked severe SCID is due to a defect in the gamma-chain of IL-2 also shared by IL-4,-7, -11 and 15, all of which are involved in lymphocyte proliferation and/or differentiation. The autosomal SCIDs arise primarily from defects in adenosine deaminase (ADA) or purine nucleoside phosphorylase (PNP) genes which results is accumulation of dATP or dGTP, respectively, and cause toxicity to lymphoid stem cells. Other genetic defects leading to
SCID include those for RAG1, RAG2 and IL-7-alpha. If suspected of SCID, the patient must not receive live vaccine, as it will result in progressing disease.
Diagnosis is based on enumeration of T and B cells and immunoglobulin measurement. Severe combined immunodeficiency can be treated with a bone marrow transplant (see MHC and transplantation). Recently, autosomal SCID patients with ADA deficiency have been treated with a retroviral vector transfected with the gene with some success.
SCID includes several disorders
Patients having both T and B cell deficiency lack recombinase activating genes (RAG1 and 2) that are responsible for the T cell receptor and Ig gene rearrangements. These patients are athymic and are diagnosed by examining the T cell receptor (TCR) gene rearrangement. Defects in B cells are not observed in early infant life because of passive antibodies obtained from the mother. NK cells are normal.
In some SCID patients, T cells may be present but functionally defective because of deficiency in signaling mediated by the CD3 chain that is associated with the TCR.
Interleukin-2 receptor common gamma chain (IL-2Rγc) may be lacking in patients there by preventing signaling by IL-2, 4, 7, 9 and 15. These patients are T and NK cell deficient.
Adenosine deaminase (ADA) is responsible for converting adenosine to inosine. ADA deficiency leads to accumulation of adenosine which interferes with DNA synthesis. The patients have defects in T, B and NK cells.
Disorders of T cells
DiGeorge’s Syndrome (Deletion 22 Syndrome)
This the most clearly defined T-cell immunodeficiency and is also known as congenital thymic aplasia/hypoplasia, or immunodeficiency with hypoparathyroidism. The syndrome is associated with hypoparathyroidism, congenital heart disease, low set notched ears and fish shaped mouth. These defects results from abnormal development of the fetus during the 6th to 10th week of gestation when parathyroid, thymus, lips, ears and aortic arch are being formed. No genetic predisposition is clear and not all DiGeorge syndrome babies have thymic aplasia. A thymic graft taken from an early fetus (13 – 14 weeks of gestation) can be used for treatment. Older grafts may result in GVH reaction. In severely immunodeficient DiGeorge patients, live vaccines may cause progressive infections.
DiGeorge syndrome is autosomal dominant (figure 2) and is caused by a deletion in chromosome 22 (figure 3). The deletions are of variable size but size does not correlate with severity of disease. In about 6% of cases, the chromosome 22 micro-deletion is inherited but most cases result from de novo deletion which may be caused by environmental factors.
T cell deficiencies with variable degrees of B cell deficiency
Ataxia-telangiectasia
Ataxia-telangiectasia is a deficiency of T cells associated with a lack of coordination of movement (ataxis) and dilation of small blood vessels of the facial area (telangiectasis). T- cells and their functions are reduced to various degrees. B cell numbers and IgM concentrations are normal to low. IgG is often reduced and IgA is considerably reduced (in 70% of the cases). There is a high incidence of malignancy, particularly leukemias, in these patients. The defects arise from a breakage in chromosome 14 at the site of TCR and Ig heavy
chain genes.
Wiskott-Aldrich syndrome
This syndrome is associated with normal T cell numbers with reduced functions, which get progressively worse. IgM concentrations are reduced but IgG levels are normal. Both IgA and IgE levels are elevated. Boys with this syndrome develop severe eczema, petechia (due to platelet defect and thrombocytopenia). They respond poorly to polysaccharide antigens and
are prone to pyogenic infection. Wiskott-Aldrich syndrome is an X-linked disorder (figure 4) due to defect in a cytoskeletal glycoprotein, CD43.
MHC deficiency (Bare leukocyte syndrome)
A number of cases of immunodeficiency have been described in which there is a defect in the MHC class II transactivator (CIITA) protein gene, which results in a lack of class-II MHC molecule on their APC. Since the positive selection of CD4 cells in the thymus depends on the presence of these MHC molecules, these patients have fewer CD4 cells and are infection prone. There are also individuals who have a defect in their transport associated protein (TAP) gene and hence do not express the class-I MHC molecules and consequently are deficient in CD8+ T cells.
Disorders of B lymphocytes
There are a number of diseases in which T cell numbers and functions are normal: B cell numbers may be low or normal but immunoglobulin levels are low. These are briefly summarized below.
X-linked infantile hypogammaglobulinemia
X-linked hypogammaglobulinemia, also referred to as Bruton’s hypoglobulinemia or agammaglobulinemia, is the most severe hypogammaglobulinemia in which B cell numbers and all immunoglobulin levels are very low. The patients have failure of B-cell maturation associated with a defective B cell tyrosine kinase (btk) gene. Diagnosis is based on
enumeration of B cells and immunoglobulin measurement.
Transient hypogammaglobulinemia
Children, at birth, have IgG levels comparable to that of the mother. Because the half life of IgG is about 30 days, its level gradually declines, but by three months of age normal infants begin to synthesize their own IgG. In some infants, however, IgG synthesis may not begin until they are 2 to 3 years old. This delay has been attributed to poor T cell help. This results in a transient deficiency of IgG which can be treated with gamma-globulin.
Common variable hypogammaglobulinemia (Late onset hypogammaglobulinemia)
These individuals have acquired deficiencies of IgG and IgA in the 2nd or 3rd decade of their life and are susceptible to a variety of pyogenic bacteria and intestinal protozoa. They should be treated with specially prepared gamma-globulin for intravenous use.
IgA deficiency
IgA deficiency is the commonest of all immunodeficiencies (1/700 of all Caucasians). About 20% of individuals with IgA deficiency also have low IgG. IgA-deficient patients are very susceptible to gastrointestinal, eye and nasopharyngeal infections. Patients with IgA deficiency have a high incidence of autoimmune diseases (particularly immune complex type) and lymphoid malignancies. Anti-IgA antibodies (IgG) are detected in 30 to 40 percent
of patients who should not be treated with γ-globulins. Laboratory diagnosis is based on IgA measurement.
Selective IgG deficiency
Deficiencies of different IgG subclasses have been found. These patients are susceptible to pyogenic infections.
Hyper-IgM immunodeficiency
Individuals with this type of immunodeficiency have low IgA and IgG concentrations with abnormally high levels of IgM. These patients cannot make a switch from IgM to other classes which is attributed to a defect in CD40L on their CD4 cells. They are very susceptible to pyogenic infection and should be treated with intravenous gamma-globulins.
NON-SPECIFIC IMMUNE SYSTEM
Primary immunodeficiencies of the non-specific immune system include defects in phagocytic and NK cells and the complement system.
Defects of the phagocytic system
Defects of phagocytic cells (numbers and/or functions) can lead to increased susceptibility to a variety of infections.
Cyclicneutropenia
This is marked by low numbers of circulating neutrophil approximately every three weeks. The neutropenia lasts about a week during which the patients are susceptible to infection. The defect appears to be due to poor regulation of neutrophil production.
Chronic granulomatous disease(CGD)
CGD is characterized by marked lymphadenopathy, hepato- splenomegaly and chronic draining lymph nodes. Leukocytes have poor intracellular killing (figure 5) and low respiratory burst. In majority of these patients, the deficiency is due to a defect in NADPH oxidase (cytochrome b558 : gp91phox, or rarely gp22phox) or other cofactor proteins (gp47phox, gp67phox) that participate in phagocytic respiratory burst. These patients can be diagnosed on the basis or poor Nitroblue tetrazolium (NBT) reduction which is a measure of respiratory burst. Interferon-gamma therapy has been successful.
Leukocyte Adhesion Deficiency
In this disease, leukocytes lack the complement receptor CR3 due to a defect in
CD11 or CD18 peptides and consequently they cannot respond to C3b opsonin.
Alternatively there may a defect in integrin molecules, LFA-1 or mac-1 arising
from defective CD11a or CD11b peptides, respectively. These molecules are involved in diapedesis and hence defective neutrophils cannot respond effectively to chemotactic signals.
Chediak-Higashi syndrome
Chediak-Higashi syndrome is marked by reduced (slower rate) intracellular killing and chemotactic movement accompanied by inability of phagosome and lysosome fusion and proteinase deficiency. Giant lysosomes (intracellular granules) are often seen (figure 6). The respiratory burst is normal. Accompanying NK cell defects and platelet and neurological disorders are noted.
DISORDERS OF COMPLEMENT SYSTEM
Complement abnormalities also lead to increased susceptibility to infections. There are genetic deficiencies of various components of complement system, which lead to increased infections. The most serious among these is the C3 deficiency which may arise from low C3 synthesis or deficiency in factor I or factor H.
I do believe all the ideas youve presented for your post They are really convincing and will certainly work Nonetheless the posts are too short for novices May just you please lengthen them a little from subsequent time Thanks for the post